 E-submission
E-submission
Articles
- Page Path
- HOME > J Liver Cancer > Volume 22(2); 2022 > Article
-
Case Report
Fibrolamellar hepatocellular carcinoma that was successfully treated with surgical resection: a case report -
Seong Kyun Na

-
Journal of Liver Cancer 2022;22(2):178-182.
DOI: https://doi.org/10.17998/jlc.2022.06.10
Published online: June 22, 2022
Department of Internal Medicine, Jeju National University College of Medicine, Jeju, Korea
-
Corresponding author: Seong Kyun Na Department of Internal Medicine, Jeju National University College of Medicine, 15 Aran 13-gil, Jeju 63241, Korea
Tel. +82-64-754-8139, Fax. +82-64-717-1131 E-mail: drcoramdeo@naver.com
Copyright © 2022 The Korean Liver Cancer Association
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 2,575 Views
- 72 Downloads
- 1 Citation
Abstract
- Fibrolamellar hepatocellular carcinoma (FLHCC) is a rare malignant hepatic cancer with characteristics that differ from those of typical hepatocellular carcinoma (HCC). Unlike conventional HCC, FLHCC is common in young patients without any underlying liver disease and is known to be associated with a unique gene mutation. This cancer type is rare in Asia, with only a few cases being reported in Korea. We report a case of FLHCC in a young woman that successfully underwent surgical resection. The efficacy of alternative treatments, such as transarterial chemoembolization or systemic chemotherapies, has not yet been established. To conclude, early diagnosis and appropriate surgical resection are important for the treatment of FLHCC.
- Fibrolamellar hepatocellular carcinoma (FLHCC) is a rare malignant hepatic cancer first reported in 1956 by Edmondson.1. Unlike classical hepatocellular carcinoma (HCC), FLHCC is common in young patients without any underlying liver disease and occurs in both women and men.2 This cancer type is rare in Asia, and only a few cases have been reported in Korea since the first case report in 1998.3-5 Since surgical resection is the only established effective treatment for FLHCC, early diagnosis and treatment are important. Here, we report a case of FLHCC in a young woman that was successfully treated by surgical resection. This case report was described according to the CARE guidelines available at https://www.care-statement.org/.
INTRODUCTION
- A 19-year-old woman without any underlying disease visited the outpatient clinic because of abnormal liver enzyme levels for the past 8 months. The patient had no chronic liver disease or viral hepatitis. She had no family history of HCC or other malignancies. She was not taking any medication, never smoked, and she drank a bottle of beer once a week. She had no abdominal pain, weight loss, or anorexia. The patient’s body mass index was 23, and there were no specific findings on physical examination. The results of the initial blood test were as follows: white blood cell count, 6,000/µL; hemoglobin, 13.8 g/dL; platelet, 438,000/µL; albumin, 4.2 g/dL; prothrombin time-international normalized ratio, 1.04; total bilirubin 0.6 mg/dL; alkaline phosphatase (ALP), 602 U/L; aspartate aminotransferase (AST), 66 U/L; and alanine aminotransferase (ALT), 106 U/L. The hepatitis B surface antigen, anti-hepatitis B surface antibody, and anti-hepatitis C virus antibody tests were all negative.
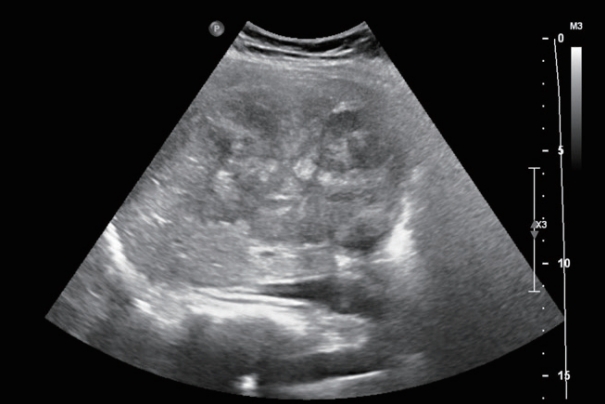
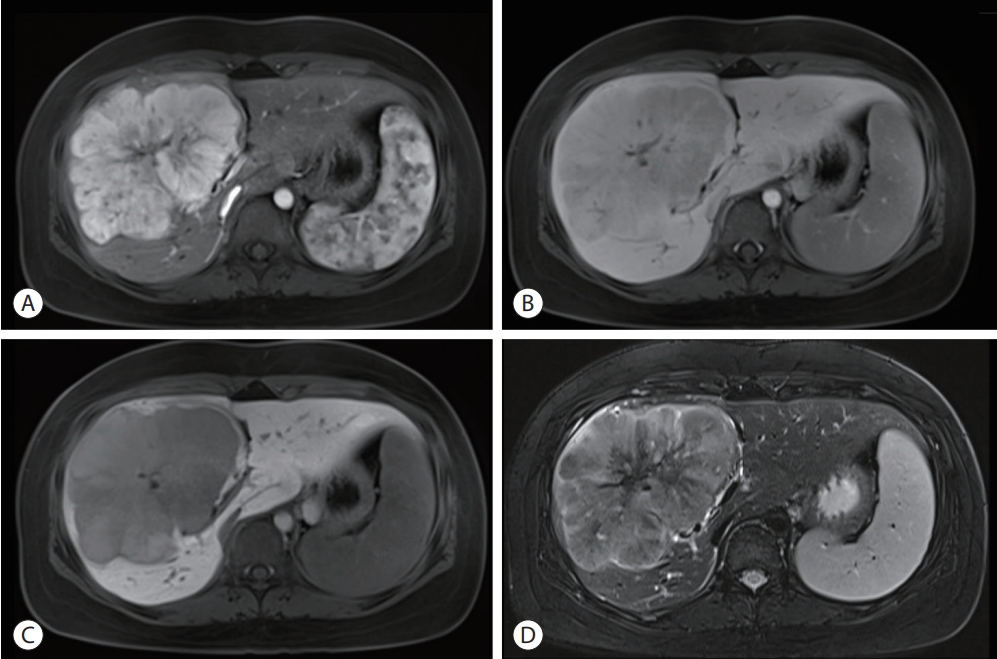
- Abdominal ultrasonography showed an approximately 13 cm-sized lobulated mass with a suspicious central scar in the right liver (Fig. 1). Magnetic resonance imaging (MRI) revealed a lobulated liver mass with a central scar, prominent arterial enhancement, and low signal intensity in the delayed and hepatobiliary phases (Fig. 2). The levels of tumor markers were as follows: alpha-fetoprotein (AFP), 3.67 ng/mL; protein induced by the absence of vitamin K or antagonist-II (PIVKA-II), 316 mAU/mL; and carbohydrate antigen 19-9, 3.51 U/mL.
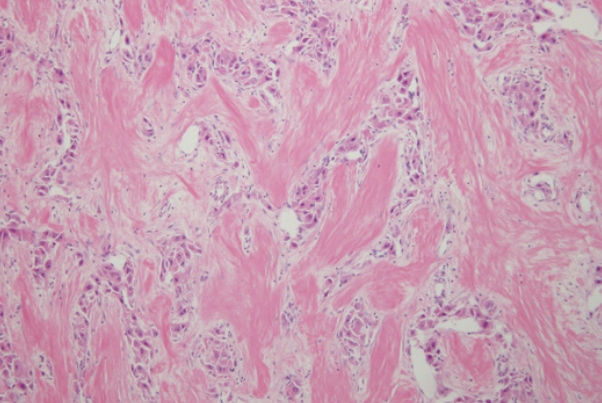
- A liver biopsy was performed for accurate diagnosis. Pathology showed large polygonal cells with abundant eosinophilic cytoplasm and fibrous stroma arranged in parallel lamellae around the tumor cells (Fig. 3). The tumor was diagnosed as a FLHCC. There was no lymph node metastasis or distant metastasis on fluorodeoxyglucose-positron emission tomography scan. The tumor, node, and metastasis stage was Ib (T1bN0M0). An extended right hemihepatectomy was performed, and the surgical specimen also demonstrated the typical histological features of FLHCC, confirming its diagnosis. After surgery, AST, ALT, and ALP levels were normalized. The PIVKA-II level decreased to 28 mAU/mL and remained within the normal range. Follow-up computed tomography (CT) was performed every 3-6 months, and there was no recurrence for 3 years and 6 months.
CASE REPORT
- FLHCC is a rare liver tumor, accounting for <1% of all primary liver cancers and 13.4% of liver cancers occurring in individuals <40 years of age.6,7 This tumor is rare in Asia, with only a few cases being reported in Korea. FLHCC has several epidemiological characteristics that differ from those of classical HCC. It tends to occur in young people <40 years of age; it does not have a male predominance8; and clear risk factors have not been identified.7 Patients usually present with nonspecific symptoms, such as abdominal pain, abdominal distension, or tumors, and may be discovered incidentally without symptoms.9 In this case, the patient did not have any specific symptoms, and the tumor was discovered by performing abdominal ultrasonography due to elevated liver enzyme levels.
- The diagnosis of FLHCC is based on clinical presentation and imaging studies. On CT, FLHCC often appears as a lobulated heterogeneously enhancing mass, and a central scar or calcification may be present. On MRI, tumors appear with hypointensity and hyperintensity on non-contrast T1- and T2-weighted images, respectively.10
- AFP, an important tumor marker of HCC, is elevated in approximately 10% of FLHCC.9,11 Another tumor marker of HCC, PIVKA-II, is elevated more frequently. In a study that analyzed FLHCC in 41 patients, PIVKA-II was elevated in all 10 patients in whom that marker was measured.12 This case also showed similar findings: AFP was normal while PIVKA-II was elevated to 316 mAU/mL.
- Pathology is the gold standard for the diagnosis. FLHCCs are characterized by large polygonal cells with abundant eosinophilic cytoplasm and abundant fibrous stroma arranged in parallel lamellae.2 The pathogenesis of FLHCC is unclear, although a unique fusion gene, DNAJB1-PRKACA (DNAJ/HSP40 homolog, subfamily B, member 1, and protein kinase, cAMP-dependent, catalytic, alpha), has been identified in nearly all FLHCC cases, and not in other HCC variants. A 400-kilobase deletion in chromosome 19 has been shown to result in a fusion gene, DNAJB1-PRKACA, which drives tumorigenesis.13 Until recently, the DNAJB1-PRKACA gene was considered to be specific for FLHCC. However, recent studies have reported that this fusion gene is also detected in other pancreatic and biliary tumors14, whereas FLHCC without the fusion gene has also been reported.15 We were not able to test this case for the DNAJB1-PRKACA alteration; however, the pathological features were typical of FLHCC and sufficient for its diagnosis. Further studies are needed to explain the roles of DNAJB1-PRKACA and other genes in the pathogenesis of FLHCC. Fibrolamellar-like features may be focally identified in otherwise conventional HCCs; such cases should not be diagnosed as FLHCC. Interestingly, HCCs with “focal fibrolamellar-like pattern” have been demonstrated to occur more commonly in older patients and to harbor BAP1 (breast cancer type 1 associated protein 1) gene abnormalities.16
- Surgical resection is the primary treatment for FLHCC. Fiveyear survival rates following surgical resection range from 58% to 82%, and the recurrence rates range from 33% to 100%.17 Approximately 20% of the patients have unresectable tumors due to metastasis or major vessel involvement.9 These patients have poor outcomes, with 5-year survival rates from 0% to 5% and a median survival of <12 months.11,18-20 Systemic chemotherapy is administered to patients with unresectable tumors. Chemotherapeutic agents include cisplatin, fluorouracil, doxorubicin, gemcitabine, and irinotecan9; however, chemotherapy has not clearly demonstrated therapeutic efficacy. Studies on sorafenib treatment are limited; in one study, 10 out of 95 patients were treated with sorafenib, eight had disease progression after 2.5-7 months, one had a mixed response followed by progression, and one was lost to follow-up.9 The efficacy of other treatments, such as transarterial chemoembolization or external radiation therapy, has not been established.9
- In this case, the persistently elevated liver enzyme levels of the patient led to an ultrasonographic evaluation, resulting in the detection of the FLHCC. Although the tumor was huge at initial diagnosis, it was successfully resected by surgery. Given the lack of effective alternative therapies, early diagnosis and surgical resection are important for the treatment of FLHCC.
DISCUSSION
-
Conflicts of Interest
The author has no conflicts of interest to disclose.
-
Ethics Statement
This study was approved by the Institutional Review Board (IRB) of Jeju National University Hospital (IRB No. 2022- 05-016), and the requirement for informed consent was waived owing to the use of preexisting medical records.
-
Funding Statement
No funding to declare.
-
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed for this case report.
-
Author Contribution
Conceptualization: SKN
Data curation: SKN
Methodology: SKN
Resources: SKN
Writing original draft: SKN
Writing review & editing: SKN
Approval of final manuscript: SKN
Article information
- 1. Edmondson HA. Differential diagnosis of tumors and tumorlike lesions of liver in infancy and childhood. AMA J Dis Child 1956;91:168−186.ArticlePubMed
- 2. Lin CC, Yang HM. Fibrolamellar carcinoma: a concise review. Arch Pathol Lab Med 2018;142:1141−1145.ArticlePubMedPDF
- 3. Kang IK, Lee JH, Lee SH, Lee JK, Lee KT, Koh KC, et al. A case of fibrolamellar hepatocellular carcinoma in Korea. Korean J Gastroenterol 1998;32:125−130.
- 4. Kim MJ, Cho EY, Choe MS, Yu ES. Fibrolamellar hepatocellular carcinoma with cytokeratin 7 expression: a case report. J Pathol Transl Med 2002;36:344−347.
- 5. Park YK, Kim YB, Jo SW, Yagn JM, Kim JK, Wang HJ, et al. Fibrolamellar hepatocellular carcinoma: a report of four cases. J Liver Cancer 2004;4:73−80.
- 6. Paradis V. Histopathology of hepatocellular carcinoma. Recent Results Cancer Res 2013;190:21−32.ArticlePubMed
- 7. El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology 2004;39:798−803.ArticlePubMed
- 8. Eggert T, McGlynn KA, Duffy A, Manns MP, Greten TF, Altekruse SF. Fibrolamellar hepatocellular carcinoma in the USA, 2000-2010: a detailed report on frequency, treatment and outcome based on the surveillance, epidemiology, and end results database. United European Gastroenterol J 2013;1:351−357.ArticlePubMedPMCPDF
- 9. Ang CS, Kelley RK, Choti MA, Cosgrove DP, Chou JF, Klimstra D, et al. Clinicopathologic characteristics and survival outcomes of patients with fibrolamellar carcinoma: data from the fibrolamellar carcinoma consortium. Gastrointest Cancer Res 2013;6:3−9.PubMedPMC
- 10. Do RK, McErlean A, Ang CS, DeMatteo RP, Abou-Alfa GK. CT and MRI of primary and metastatic fibrolamellar carcinoma: a case series of 37 patients. Br J Radiol 2014;87:20140024. ArticlePubMedPMC
- 11. Stipa F, Yoon SS, Liau KH, Fong Y, Jarnagin WR, D'Angelica M, et al. Outcome of patients with fibrolamellar hepatocellular carcinoma. Cancer 2006;106:1331−1338.ArticlePubMed
- 12. Pinna AD, Iwatsuki S, Lee RG, Todo S, Madariaga JR, Marsh JW, et al. Treatment of fibrolamellar hepatoma with subtotal hepatectomy or transplantation. Hepatology 1997;26:877−883.ArticlePubMedPMC
- 13. Graham RP, Jin L, Knutson DL, Kloft-Nelson SM, Greipp PT, Waldburger N, et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod Pathol 2015;28:822−829.ArticlePubMedPDF
- 14. Vyas M, Hechtman JF, Zhang Y, Benayed R, Yavas A, Askan G, et al. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod Pathol 2020;33:648−656.ArticlePubMedPMCPDF
- 15. El Dika I, Bowman AS, Berger MF, Capanu M, Chou JF, Benayed R, et al. Molecular profiling and analysis of genetic aberrations aimed at identifying potential therapeutic targets in fibrolamellar carcinoma of the liver. Cancer 2020;126:4126−4135.ArticlePubMedPMCPDF
- 16. Hirsch TZ, Negulescu A, Gupta B, Caruso S, Noblet B, Couchy G, et al. BAP1 mutations define a homogeneous subgroup of hepatocellular carcinoma with fibrolamellar-like features and activated PKA. J Hepatol 2020;72:924−936.ArticlePubMed
- 17. Mavros MN, Mayo SC, Hyder O, Pawlik TM. A systematic review: treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma. J Am Coll Surg 2012;215:820−830.ArticlePubMed
- 18. Kakar S, Burgart LJ, Batts KP, Garcia J, Jain D, Ferrell LD. Clinicopathologic features and survival in fibrolamellar carcinoma: comparison with conventional hepatocellular carcinoma with and without cirrhosis. Mod Pathol 2005;18:1417−1423.ArticlePubMedPDF
- 19. Moreno-Luna LE, Arrieta O, García-Leiva J, Martínez B, Torre A, Uribe M, et al. Clinical and pathologic factors associated with survival in young adult patients with fibrolamellar hepatocarcinoma. BMC Cancer 2005;5:142. ArticlePubMedPMCPDF
- 20. Ramai D, Ofosu A, Lai JK, Gao ZH, Adler DG. Fibrolamellar hepatocellular carcinoma: a population-based observational study. Dig Dis Sci 2021;66:308−314.ArticlePubMedPDF
References
Figure & Data
References
Citations
- Fibrolamellar Hepatocellular Carcinoma (FLHCC) in a Young Patient Presenting With Nausea and Vomiting After a Greasy Meal
Mohamed Ismail , Sahiba Singh, Menna-Allah Elaskandrany , David s Kim, Yazan Abboud, Michael Bebawy, Abena Oduro, Ritik mahaveer Goyal, Omar Mohamed , Weizheng Wang
Cureus.2024;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Download Citation
Download Citation
- Download Citation
- Close
- Related articles
-
- The development of hepatocellular carcinoma during long-term treatment for recurrent non-small cell lung cancer: a case report
- Multidisciplinary approach for hepatocellular carcinoma arising from cirrhotic liver with Budd-Chiari syndrome: a case report
- Hepatocellular carcinoma with Budd-Chiari syndrome due to membranous obstruction of the inferior vena cava with long-term follow-up: a case report
- Hepatocellular carcinoma diagnosed in a patient who had Fontan operation 30 years ago: a case report
- Long-term survival after CCRT and HAIC followed by ALPPS for hepatocellular carcinoma with portal vein invasion: a case report

 Follow JLC on Twitter
Follow JLC on Twitter