Deciphering and Reversing Immunosuppressive Cells in the Treatment of Hepatocellular Carcinoma
Article information

Abstract
Use of immune checkpoint inhibitors (ICIs) in hepatocellular carcinoma (HCC) has been partially successful. However, most HCC patients do not respond to immunotherapy. HCC has been shown to induce several immune suppressor mechanisms in patients. These suppressor mechanisms include involvement of myeloid-derived suppressor cells, regulatory T-cells, functionally impaired dendritic cells (DCs), neutrophils, monocytes, and tumor associated macrophages. The accumulation of immunosuppressive cells may lead to an immunosuppressive tumor microenvironment as well as the dense fibrotic stroma which may contribute to immune tolerance. Our laboratory has been investigating different cellular mechanisms of immune suppression in HCC patients. In vitro as well as in vivo studies have demonstrated that abrogation of the suppressor cells enhances or unmasks tumor-specific antitumor immune responses. Two or three effective systemic therapies including ICIs and/or molecular targeted therapies and the addition of innovative combination therapies targeting immune suppressor cells may lead to increased immune recognition with a greater tumor response. We reviewed the literature for the latest research on immune suppressor cells in HCC, and here we provide a comprehensive summary of the recent studies in this field.
Background
Studies in the last decade have increasingly recognized the anti-tumor function of the immune system as a fundamental principle of the malignant process as well as a new target for cancer treatment. While many immune modulating therapies are of significant scientific and clinical interest, immune checkpoint inhibitors (ICIs) have shown impressive clinical efficacy. Currently, there are two classes of ICIs receiving significant clinical attention. The first class includes inhibitors of the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) ligand. The second class includes inhibitors of the programmed cell death protein 1 (PD-1). With the recent FDA approval of nivolumab and pembolizumab for the treatment of hepatocellular carcinoma (HCC) patients who have been previously treated with sorafenib, immunotherapy has become a treatment option for patients with HCC despite recent results from two negative trials. We and other research groups have previously demonstrated that spontaneous tumor-specific immune responses occur frequently in HCC patients. Both humoral and cellular tumor-specific immune responses to HCC has been reported [1-5]. Immune-based approaches for treatment of HCC are aimed primarily at immune effector cell functions rather than the tumor immune microenvironment, which hosts different immune cells with immunosuppressive functions. The tumor microenvironment (TME) has also been shown to influence the response to immunotherapies, including ICIs. Accumulation of immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), regulatory T-cells (Tregs), tumor-associated macrophages (TAMs) and neutrophils have been shown to limit the response to immunotherapies and also correlate with a poor prognosis [6-8].
Tumors are broadly classified into categories of infiltrated-excluded, infiltrated-inflamed, or infiltrated with tertiary lymphocyte structures [9]. Infiltrated-inflamed tumors are the most susceptible to immune checkpoint therapy. Treatment with antibodies against CTLA-4 lead to a T-cell–enriched tumor phenotype [10]. An extensive analysis of 10,000 tumors, comprising 33 diverse cancer types (including HCC), showed six immune subsets of cancers [11]. Most HCCs were characterized as lymphocyte depleted; tumors of high-macrophage or inflammatory subtypes (high ratio of Th1 to Th2 cells) had a large number of Th17 cells and a balanced ratio of macrophages to lymphocytes [12]. Technologies such as RNA sequencing combined with CIBERSORT [13] and XCell [14], in combination with mass spectrometric analysis of single cells [15], allow for multiplex high-dimensional analysis of immune cell populations in tumors.
These studies also suggest that tumor-specific cellular immune responses can be potentially overshadowed by different suppressor mechanisms, disabling effective anti-tumor immunity. In this article, we review and summarize the current knowledge on cellular suppressor mechanisms in patients with HCC.
IMMUNO SUPPRESSIVE CELLS
1. Myeloid-derived suppressor cells
MDSCs are a mixture of immature myeloid cells, immature granulocytes, monocytes, macrophages, DCs, and myeloid progenitor cells generated in the context of cancer [16-19]. Pathological activation is the result of persistent stimulation of the myeloid compartment with relatively low-strength signals coming from the tumors or sites of chronic inflammation. Myeloid cells generated under these conditions are poorly phagocytic, produce high levels of reactive oxygen and nitrogen species, and predominantly anti-inflammatory cytokines [20]. As a result, these cells are not able to effectively perform the normal functions of myeloid cells and acquire potent immune-suppressive potential. Although MDSCs suppress diverse immune functions, their main immunosuppressive actions are exerted through the inhibition of T-cells and natural killer (NK) cells and induction of Tregs. The major factors involved in MDSC-mediated immunosuppression include arginase-1 (ARG1), inducible nitric oxide synthase (iNOS), reactive oxygen species (ROS), transforming growth factor-beta (TGF-β), interleukin-10 (IL-10), COX2, and indoleamine 2,3-dioxygenase (IDO) [21,22]. ARG1 depletes L-arginine and leads to cell cycle arrest in the tumor-infiltrating T-cells at G0-G1 phase. Depleted L-arginine increases NO production by iNOS, resulting in increased ROS production; all result in the down-regulation or desensitization of the T-cell receptor and induction of T-cell anergy. IDO degrades L-tryptophan and leads to the suppression of T- and NK cells, and activation of Tregs [22]. Studies have also suggested that the immunosuppressive mechanisms of MDSCs may vary at different sites. In peripheral lymphoid structures, polymorphonuclear (PMN)-MDSCs (CD11b+Ly6G+Ly6Clo) have a high level of ROS production and suppress T-cells function in an antigen-specific manner. In contrast, monocytic (M)-MDSCs (CD11b+Ly6G–Ly6Chi) suppress not only antigen-specific but also nonspecific T-cell responses by expressing various factors such as ARG1, NO, TGF-β, and IL-10 [23]. In TME, because of hypoxia, ROS levels in PMN-MDSCs are substantially reduced; however, the levels of ARG1 and other factors responsible for nonspecific T-cell suppression show an increase [21]. Additionally, MDSCs influence TME by inducing tumor angiogenesis through the production of several angiogenic factors and vascular-modulating enzymes [24]. For example, bombina variegata peptide 8 (Bv8, a homolog of endocrine-gland-derived vascular endothelial growth factor [VEGF]), produced by MDSCs through granulocyte colonystimulating factor (G-CSF)-dependent signal transducer and activator of transcription 3 (STAT3) signaling, was demonstrated to promote angiogenesis and hematopoietic cell mobilization [25]. Accumulation of MDSC in TME was associated with tumor refractoriness to anti-VEGF treatment; anti-G-CSF therapy or anti-Bv8 therapy could enhance the responsiveness of anti-VEGF treatment [26,27]. Moreover, matrix metalloproteinase-9 (MMP-9)-expressing CD11b+ myelomonocytic cells have been shown to be critical for the formation of tumor vasculature. Tumor growth could be inhibited in MMP-9 knockout mice or by the deletion of MMP-9 in CD11b+Gr1+ MDSCs [28,29]. In addition, MDSCs could acquire endothelial cell properties in TME and directly get incorporated into the tumor endothelium [29].
Recent studies have investigated the roles of MDSCs in the efficacy of ICIs in mouse HCC models. Chiu et. al studied multiple orthotopic mouse HCC models and found tumor hypoxia induced the ectoenzyme, ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2), by stabilizing hypoxia-inducible factor-1 (HIF-1) in cancer cells. ENTPD2 supported the maintenance of MDSCs, and targeting ENTPD2 inhibited tumor growth and enhanced the efficacy of PD-1/CTLA-4 blockade [30]. Zhou et al. demonstrated that the overexpression of cell cycle-related kinase (CCRK), a cyclin dependent kinase family member, increased MDSC accumulation and T-cell suppression in liver-specific CCRK inducible transgenic mice [31]. Targeting CCRK or downstream IL-6 signaling reduced tumor-infiltration of MDSCs and increased intra-tumoral immunity via interferon (IFN)-γ+ tumor necrosis factor-α (TNF-α)+CD8+ T-cells. As CCRK targeting drugs are not on the market, it may be easier to block IL-6 and combine this treatment with ICIs. However, more preclinical studies are needed to confirm this combination strategy [32].
In human peripheral blood mononuclear cell (PBMC), the equivalent to PMN-MDSC are defined as CD11b+CD14–CD15+ or CD11b+CD14–CD66b+ and M-MDSC as CD11b+CD14+HLA-DRlow/negCD15–. CD33 myeloid marker can be used instead of CD11b because very few CD15+ cells are CD11b– [23]. Recently, lectin-type oxidized LDL receptor-1 (LOX-1) has been identified as a new marker for PMN-MDSCs in humans, further facilitating the discrimination of human PMN-MDSCs from mature neutrophils [33]. We have identified human monocytic CD14+HLA-DRlow/neg MDSCs in patients with HCC and described an increase in the frequency of CD14+HLA-DRlow/neg MDSCs in peripheral blood and ascites in these patients [34]. We showed that CD14+HLA-DRlow/neg MDSCs failed to induce proliferative T-cell responses and did not mature into DCs in vitro . Apart from their ability to suppress nonspecific T-cell responses, MDSCs also masked alfa-fetoprotein (AFP)-specific T-cell responses [34]. In addition, we showed that human MDSCs induced a T regulatory phenotype when co-cultured with CD4+ T-cells [34]. Interestingly, while MDSCs induced forkhead or winged helix family of transcription factor P3 (FoxP3)+ Tregs, CD14+HLA-DR+ cells induced a different T helper subtype, Th17 cells [35].
To better understand the biology and the clinical relevance of human MDSCs, we further examined the interaction between MDSCs and other immune cells like NK and cytokineinduced killer (CIK) cells. NK cells represent an important cell type in the context of HCC. NK cells are impaired in function in HCC patients [36]. We have previously demonstrated that MDSCs are potent suppressors of NK cells in patients with HCC [37]. In addition, we have demonstrated that adoptive cell transfer of CIK cells into tumor bearing mice induced inflammatory mediators (e.g., CX3CL1, IL-13) in the TME and an increase of tumor infiltrating MDSCs leading to impaired anti-tumor activity in two different murine HCC tumor models [38]. MDSCs efficiently suppressed the cytotoxic activity of CIK cells in vitro . Tadalafil treatment, a phosphodiesterase- 5 (PDE5) inhibitor, has previously been shown to reverse MDSC suppressor function via ARG1 and iNOS blockade [39,40]. We showed that systemic treatment with PDE5 inhibitor prevented MDSC accumulation in the TME upon murine CIK cell therapy and increased its anti-tumor effica cy. Similar results were observed when human CIK cells were tested in vitro in the presence of CD14+HLA-DRlow/neg MDSCs. Treatment of MDSCs with a PDE5 inhibitor suppressed MDSCs suppressor function and enhanced CIK activity against human HCC cell lines in vitro [38].
2. Regulatory T-cells
CD4+CD25+ Tregs are a minor but functionally unique population of T-cells which maintain immune homeostasis in immune tolerance and the control of autoimmunity. In vitro Tregs can inhibit immune responses mediated by both CD4+ and CD8+ effector T-cells by a contact-dependent and cytokine-independent mechanism [41-43]. However, the mechanism of immune suppression is much more complex in vivo [16,44], and includes events such as IL-2 depletion by CD25 (IL-2 receptor), competition between CD28 and CTLA-4, CTLA-4-mediated down-regulation of CD80 and CD86 [45], and expression of TGF-β and IL-10 [46]. The recruitment of Tregs in HCC occurs via the CCR6-CCL20 axis [47] and CCL22 induction by tumor cell-secreted IL-1α [48]. FoxP3 is not only essential for development of Tregs but also remains the best marker to identify these cells. However, several studies have shown that activation of human non-Tregs can also lead to expression of FoxP3 in vitro , suggesting that this marker needs to be used with caution [17]. Alternatively, it has been suggested that analysis of FoxP3 methylation status can be used to determine the presence of Tregs in humans [49]. In addition, FoxP3 up-regulation and conversion of CD4+ T-cells into Tregs may be fostered by poor stimulation of naive CD4+ T-cells combined with TGF-β signaling by tumor cells. Previous studies have demonstrated the pivotal role of Tregs in tumor immunology and their ability to suppress anti-tumor immune responses. Accordingly, targeting Tregs has been shown to boost anti-tumor immunity. Involvement of CD4+CD25+ Tregs in human cancer has been observed in peripheral blood and tumor tissues from patients with several types of cancer [50-52]. We and others have been able to demonstrate that Tregs are increased in peripheral blood and tumor-infiltrating lymphocytes of patients with HCC [8,53,54]. While initial investigations only demonstrated an increase in Tregs frequencies in patients with HCC, follow-up studies have explored a potential correlation with disease progression and patients’ outcomes [55,56]. One study demonstrated that an up-regulation of Tregs was associated with a significantly reduced CD8+ T-cell infiltration of tumors [57]. Recently, deep single-cell RNA sequencing on 5,063 single T-cells isolated from peripheral blood, tumor, and adjacent normal tissues from six HCC patients revealed that exhausted CD8+ T-cells and Tregs were preferentially enriched and potentially clonally expanded [58]. Layilin was up-regulated on Tregs and repressed the CD8+ T-cell functions in vitro [58]. A correlation between poor survival and an increase in Tregs was also shown in the same report and validated by other studies [59,60]. Finally, patients with advanced HCC had a higher percentage of intra-hepatic CD8+FoxP3+ Tregs than did patients with early disease, suggesting that CD8+FoxP3+ Tregs also represent another immune escape mechanism [61].
As Tregs are increased in patients with HCC and correlate with a worse outcome, we examined whether Tregs also suppress tumor-specific T-cell responses. We showed that in vitro depletion of Tregs unmasks AFP-specific immune responses in PBMC isolated from patients with HCC. Based on this in vitro observation, we performed a clinical trial targeting Tregs in patients with HCC. Patients were treated with low-dose cyclophosphamide, which had been shown in mice to target Tregs. While the number of patients treated in this study was too small to draw any definite conclusions, the results demonstrated that the frequency of Tregs in peripheral blood can be temporarily reduced by low-dose cyclophosphamide treatment [44,62].
3. Tumor-associated macrophages
Macrophages are a major component of the leukocyte infiltrate that is present, to a widely varying extent, in all tumors [63]. Dissection of the roles of TAMs in tumor progression has elucidated the contributions of other inflammatory cells and mediators, such as inflammatory cytokines. In fact, TAMs have a dominant role as orchestrators of cancer-related inflammation.
Liver macrophages consist of ontogenically distinct populations, namely, the resident Kupffer cells (KCs) and monocyte- derived macrophages (Mo-Mfs). KCs are self-renewing and nonmigratory phagocytes. In the TME, chemokines secreted by malignant and stromal cells recruit bone marrow-derived Ly6Chi monocytes. These infiltrating monocytes subsequently give rise to a large number of Mo-Mfs, which further differentiate and can replace and acquire a phenotype that is almost indistinguishable from resident KCs under specific circumstances [64-69]. After infiltration, Mo-Mfs seem to acquire the ability to proliferate [66]. It is however, unclear if they are able to sustain the number of TAMs in tumor lesions independently when not being continually recruited. As a result of this continuous transition, the compartment of hepatic myeloid cells consists of subtypes of macrophages in a different stages of differentiation. Each state is associated with stereotypic alterations in cell surface marker expression, which can be used for identification. In many studies, CD68 is used as an indicator for tissue macrophages; however, this marker is not sufficiently specific. More recently, two markers were proposed to distinguish between Mo-Mfs and KCs. Clec4F and Tim4 are expressed by KCs but absent from infiltrating Mo-Mfs [70]. Additionally, these markers can be used to discriminate between KCs and recently differentiated Mo-KCs as the latter do not express Tim4 during the first week, post-differentiation. However, with time, Mo-KCs will also gain expression of Tim4 [66,67]. It is not clear to what extent TAMs are derived from tissue-resident liver cells or only represent infiltrating bone marrow derived Mo-Mfs. Although KCs were initially thought to be only involved in antitumor immunity, there is substantial evidence suggesting that KCs are part of the TAM population and enhance tumor progression [71-74].
Defining TAMs as a single population has limitations due to an overgeneralized definition of TAMs and the need for further subdivision according to their polarization. Macrophages can be classified into a classically activated (pro-inflammatory) M1 state triggered by interferon-γ and/or lipopolysaccharide, or an alternatively activated (anti-inflammatory) M2 state induced by IL-4 [70]. This pro- and anti-inflammatory paradigm leads to the confusing assumption that in an inflammation-related tumor, an M2 phenotype would be beneficial. However, during tumor progression in HCC, macrophage function is skewed from M1 to M2 phenotype [75,76]. The polarization of macrophages not only depends on the disease stage but also differs between tumoral nodules or within different areas of the same tumor. In human HCC, for example, most of the perivascular macrophages are more M1-like compared to the M2-like TAM in hypoxic areas [64,77]. M2 macrophages are characterized by producing high levels of IL-10 that induce Tregs expansion and impairs NK cell activation [78]. In addition, TAM promote tumor angiogenesis and dissemination [79,80]. A distinct subset of monocytes expressing TIE2 with enhanced pro-angiogenic properties has been described in peripheral blood and in tumor infiltrate [81-83].
4. Tumor-associated neutrophils
Tumor-associated neutrophils (TANs) have been proposed to support tumor development by promoting cellular transformation, tumor progression, and antitumor immunity. Proinflammatory cytokine IL-17 is a critical mediator for the recruitment of neutrophils in the TME [84]. TANs may influence tumor progression through recruiting macrophages and Tregs to the TME, the paracrine release of cytokines and chemokines with protumor or antitumor functions, depending on the TME [85]. CCL2 and CCL17 are the most highly expressed chemokines in TANs and peripheral blood neutrophils activated by HCC cells [85]. CCL2 and CCL17 recruited CCR2+ macrophages and CCR4+ Tregs in vitro , respectively [85]. TANs infiltration has been described to positively correlate with angiogenesis progression at the tumor-invading edge of HCC patients and to be a poor prognostic factor [86,87].
5. Monocytes
The role of monocytes in the TME of HCC has been thoroughly studied by Zheng’s research group. It has been shown that expression of PD-L1 on the surface of monocytes and macrophages in the peritumoral stroma suppresses T-cell responses [88]. They also demonstrated that monocytes not only suppressed T-cell function directly but also induced Th17 cells [89] as well as IL-17-secreting effector CD8+ T-cells (Tc17 cells) [90].
6. Th17 Cells
It has been shown that Th17 cells are associated with poor outcomes in HCC [91]. We have shown an increase in the frequency of Th17 cells in peripheral blood and Th17-related cytokines (IL-17, IL-23) in tumor supernatants from HCC patients. This observation prompted us to investigate whether Th17 cells have an effect on CD8+ T-cell function. In vitro studies demonstrated that Th17 cells inhibit IFN-γ production and proliferation by CD8+ T-cells. Further analysis revealed that only the CCR4+CCR6+ subpopulation of Th17 cells were responsible for this effect, which prompted us to further examine CCR4+CCR6+ Th17 cell populations in HCC patients. Interestingly, we found only an increase in CCR4+CCR6+ Th17 cells in peripheral blood from patients with HCC but not in CCR4negCCR6+ Th17 cells [92]. CCR4+CCR6+CD4+ T-cells demonstrated a marked suppression of CD8+ T-cell responses in contrast to CCR4negCCR6+CD4+ T-cells.
7. Hepatic sinusoidal endothelial & stellate cells
In the liver, it has been reported that sinusoidal endothelial cells induce immune tolerance against CD8+ T-cells against tumor-associated antigens (TAAs) released from cancer cells that have undergone apoptosis [93]. In addition, sinusoidal endothelial cells have been reported to contribute to the immunosuppressive environment in the liver by inducing Tregs or PD-L1 through membrane-bound TGF-β [94]. Furthermore, liver stellate cells are present in the liver. In HCC patients in which the cells are activated, an immunosuppressive environment for the tumor is induced and has been reported to have a poor prognosis [95]. Activated stellate cells have been reported to induce monocytes to an immunosuppressive phenotype, MDSCs, T-cell dysfunction, and apoptosis via PD-L1 expression [96].
STRA TEGIES FOR TARG ETING IMMUNO - SUPPRESSIVE CELLS (Table 1)

Strategies for targeting immunosuppressive cells
1. MDSCs: MDSCs inhibition could be a useful adjunct to immune therapies and can be placed into five categories [97]
1) Deactivation of MDSC by PDE5 inhibitors such as sildenafil and tadalafil via the degradation of cyclic guanosine monophosphate (cGMP) leading to reduction in ARG1 and NOS2 expression [40]. STAT3 is a critical transcription factor for immunosuppressive activity and proliferation of MDSCs. A STAT3 oligonucleotide inhibitor, danvatirsen (AZD9150), was tested in a phase I/Ib clinical trial of patients with advanced/metastatic HCC (NCT01839604). Thirty-nine patients received the study agent in the escalation or expansion cohort. Only one patient in the escalation cohort had a partial response. The most common adverse events were transaminase elevation and thrombocytopenia. Histone deacetylase (HDAC) inhibitors may suppress MDSC function by reducing ARG1, iNOS, and COX-2 levels [98]. Several clinical trials have tested various HDAC inhibitors in HCC [99,100]. Although these agents were generally tolerated, the effect of such therapy on MDSCs was not evaluated.
2) Differentiation of MDSC into mature cells by using all-trans-retinoic acid (ATRA) [101] or a derivative of vitamin A [102].
3) Blocking development of MDSC by N-Bisphosphonate via decreased prenylation of MMP9 which may influence MDSC generation/function by cleaving c-kit, which is believed to play a role in MDSC mobilization from the bone marrow niche [103].
4) Depletion of MDSC by cytotoxic agents like gemcitabine [104], cisplatin, paclitaxel, or 5-fluorouracil (5-FU) or heat shock protein 90 (HSP90) inhibitor, 17-dimethylaminoethylamino-17-demethoxygeldanamycin. A preclinical study demonstrated that cabozantinib which showed significant survival benefits compared to a placebo in patients with HCC who had been previously treated with sorafenib [105] reduced intra-tumoral PMN-MDSCs and enhanced the therapeutic effect of ICIs in a prostate cancer model [106]. A recent preclinical study demonstrated that MDSCs could be selectively targeted by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor 2 (TRAIL-R2/DR5) agonist [107]. A phase I clinical trial testing the agonistic TRAIL-R2 antibody DS-8273a in patients with advanced cancer, including HCC, found that DS-8273a eliminated MDSCs without affecting mature myeloid or lymphoid cells; the decrease in MDSCs was associated with progression-free survival [108].
5) Blockade of MDSC trafficking into TME is crucial for their main immunosuppressive function to be manifested. Therefore, inhibiting chemokine receptors may reduce the number of MDSCs in TME. Chemokine receptor CCR2 and the interaction of its ligand CCL2 are required not only for the recruitment of M-MDSCs and TAMs but also for their suppressive function [109,110]. CCR5 is another chemokine receptor that is expressed in many immune cells. The CCR5-CCR5 ligand axis was found to be critical for the mobilization of PMN-MDSCs [111]. However, there has been no clinical development of inhibitors of these chemokine receptors in HCC. Recently, our group demonstrated that tadalafil, a PDE5 inhibitor prevented MDSC accumulation in the TME by decreasing inflammatory cytokines/chemokines (e.g., CX-3CL1, IL-13), both in subcutaneous and orthotopic murine HCC models [38].
2. Tregs: Methods to target Tregs include depletion of Tregs, blocking immune checkpoint receptors, recruitment of Tregs, and treatment of cells with inhibitory cytokines
1) Depletion of Tregs: CD25 is a well-known Treg cell marker and was found to be preferentially expressed in tumors in vivo [112], and depletion of CD4+CD25+ Tregs using anti-CD25 monoclonal antibodies was capable of enhancing CCL21-mediated antitumor immunity in a mouse HCC model [113]. Treatment with Fc-optimized anti-CD25 antibody resulted in the effective depletion of Tregs and an increase in the effector T-cells (Teff)-to-Tregs ratio, leading to tumor regression and increased survival [112].
2) Immune Checkpoint Inhibitors: Mechanistically, anti-CTLA-4 was first thought to prevent Tregs from intercepting costimulatory signals from DCs, resulting in DC-induced Teff cell activation and proliferation. Ipilimumab and tremelimumab induce significant activation and expansion of Teff and CD8+ T-cells [114-118]. The effect of ipilimumab was recently substantiated by depleting Tregs via antibody-dependent cell-mediated cytotoxicity (ADCC) [119]. However, tremelimumab, which does not have ADCC activity, had a similar therapeutic effect, suggesting that Treg depletion may not be the main mechanism of action of ipilimumab. Another Tregspecific marker glucocorticoid-induced TNFR family related gene (GITR) is also a target for tumor infiltrating Tregs. Unlike in Tregs, GITR acts as a costimulatory molecule in Teff cells, suggesting a beneficial effect in cancer therapy. In animal models, anti-GITR antibody induced antitumor activity by increasing Teff cells [120]. Combined treatment with anti-GITR and anti-PD-1 antibodies further enhanced antitumor activity in some HCC patients [121]. OX40, a member of the TNF receptor family, has a mechanism of action similar to that of GITR; that is, anti-OX40 antibody stimulates Teff cells but inhibits Tregs. High-OX40 expression was associated with the activation of multiple immunosuppressive pathways and poor prognosis in HCC patients [122]. Anti-OX40 antibody enhanced CD8+ T-cell-mediated antitumor immunity in animal models of cancer [123]. Antibodies against GITR and OX40 are now in clinical trials [124]. Combining Treg cell depletion with ICIs resulted in a synergistic effect in an animal model of Claudin-low breast cancer, a subtype of triple-negative breast cancer [125]. Tregs depletion and ICIs each had little effect on tumor growth, whereas their combination greatly reduced tumor burden [125].
3) Blocking Tregs Recruitment: Infiltration of Tregs into tumors is a prerequisite for their activity. Tregs express a variety of chemokine receptors, including CCR4, CCR5, CCR6, CCR7, CCR10, CXCR3, and CXCR4, and migrate efficiently in response to tumor-derived chemokines [126-128]. CCR4 is preferentially expressed on tumor infiltrating Tregs rather than on Teff cells [129], with the CCL17/22-CCR4 axis playing an important role in multiple cancers including HCC [126-128,130]. A monoclonal antibody targeting CCR4 has shown promising results, effectively depleting Tregs, both in vitro and in clinical trials in human cancer patients [128,131]. CXCR3+ Tregs selectively accumulate in ovarian cancer and block the interactions between CXCR3 and its ligands CXCL9, CXCL10, and CXCL11, thereby suppressing tumor growth [132].
4) Blocking Inhibitory Cytokines: Because the TME is rich in immunosuppressive cytokines that strengthen the activity of Tregs, neutralizing these cytokines may reestablish effective antitumor immunity. Genetic ablation or blocking of IL-10 or TGF-β signaling results in tumor regression [133-136]. Overexpression of IL-35 was correlated with CD39+FoxP3+ Tregs infiltration and HCC aggressiveness and was an independent prognostic factor [137]. In addition, neutralization of IL-35 or Treg-specific deletion of IL-35 was found to enhance antitumor T-cell responses and reduce tumor growth in various mouse tumor models [138]. Interestingly, IL-35 produced by Tregs promoted the expression of several inhibitory molecules, including PD-1, TIM-3, and LAG-3, leading to T-cell exhaustion. The higher numbers of IL-35-expressing Tregs present in tumors than in the spleen can be exploited for tumor-specific blockade of Treg cell function without affecting Treg function in general [138].
3. TAMs: Current approaches for TAMs-targeted therapy are aimed at decreasing the population of TAMs by eliminating TAMs present in the tumor, blocking recruitment of bone marrow-derived monocytes, and/or reprogramming TAM polarization to anti-tumoral behavior
1) Depletion of TAMs: Administration of liposome-encapsulated clodronate partially depleted TAMs, resulting in reduced tumor growth in a murine Hepa1-6 cell-transplanted tumor model. Not only was the total amount of TAMs reduced but, in addition, the number of M2-like TAMs in tumors of liposome-treated mice was found to be significantly lower than that in tumors of untreated mice. In contrast, the number of M1 TAMs was not significantly affected. According to the authors, these results suggest that after depleting the majority of TAMs, the remaining macrophages might undergo a phenotypical transition [139].
2) Inhibiting Recruitment of Monocytes: The chemokine C-C motif ligand 2 (CCL2, also referred to as monocyte chemoattractant protein 1 or MCP-1) and the corresponding CCL2-CCR2 signaling axis are important targets to inhibit the recruitment of monocytes. Treatment with a CCR2 antagonist inhibited HCC tumor growth in different murine models. The therapy reduced the infiltration of blood Ly-6Chigh inflammatory monocytes, subsequently lowered the number of TAMs in the HCC lesions, and reduced most of the cytokines or chemokines produced by M2-like TAMs (CD206-positive cells). Moreover, the reduced number of remaining TAMs shifted towards the M1 phenotype. The CCR2 antagonist also supported tumor-infiltrated CD8+ T-cells by blocking TAM-mediated immunosuppression [140,141]. In addition, Teng et al. showed the tumor-inhibiting effect of a CCL2 neutralizing antibody by reducing the population of inflammatory myeloid cells in an HCC mouse model [142].
3) Reprogramming Polarization of TAMs: Oral administration of baicalin, a natural flavonoid present in several medicinal plants, inhibited growth of HCC lesions in an orthotopic mouse model by initiating TAM reprogramming to an M1-like phenotype with proinflammatory cytokine production. Coculturing of HCC cells with baicalin-treated macrophages resulted in reduced proliferation and motility in vitro [143]. Colony-stimulating factor-1 (CSF-1) and its receptor, CSF-1R, regulate the differentiation and function of macrophages. CSF-1R blockade by a competitive inhibitor significantly delayed tumor growth in murine xenograft models. The compound inhibited the proliferation of macrophages in vitro ; however, macrophage infiltration was not decreased in vivo . Thus, the effect is not mediated by TAM depletion. Gene expression profiling showed that TAMs in the treated tumors are polarized towards an M1-like phenotype [144].
4) Blocking the Downstream Effect of TAMs: TAMs represent a major paracrine IL-6 source during HCC progression, and autocrine IL-6 contributed significantly to HCC initiation from HCC progenitor cells. Blockade of IL-6 signaling using tocilizumab, an anti-IL-6 receptor antibody approved by the FDA for the treatment of rheumatoid arthritis, was able to inhibit TAM-stimulated activity of cancer stem cells in vitro and in vivo [145].
5) Therapy Affecting TAMs with Currently Used Clinical Therapies: Lenvatinib has been reported to enhance the therapeutic effect of ICIs by reducing TAMs locally at the tumor and enhancing antitumor IFN signal [146]. In fact, also in human clinical trials, the efficacy of the combination therapy of lenvatinib and pembrolizumab has been reported [147]. The efficacy of the combination of VEGF inhibitor (bevacizumab) and anti-PD-L1 antibody (atezolizumab) for HCC has been reported [147]. Because VEGF increases TAMs and Tregs and enhances the expression of immune checkpoint molecules including PD-1 molecules of CD8+ T-cells [148,149], combination therapy of VEGF inhibitors and anti-PD-1 antibodies makes sense. It is expected that multiplex immunotherapy combining such molecular targeted drugs with immunotherapy will be increasingly developed in the future.
CONCLUSIONS
HCC has unique immune evasive microenvironment, in which multiple cellular and molecular immune evasive mechanisms could be targeted by immunotherapy. Targeting immune checkpoint molecules such as CTLA-4, PD-1, and PD-L1 has opened new opportunities for cancer treatment with successful responsive outcomes. Emerging evidences in HCC patients suggest that modulation of the immune checkpoint pathways could lead promising clinical responses with manageable toxicity profile. However, we believe that it will be important in the future to test treatment modalities which will target immunosuppressive cell populations, especially in trials which aim to enhance anti-tumor immune responses (Fig. 1). Future approaches for HCC immunotherapy are to establish novel strategies rationally combined with other current or future treatment options in order to maximize the antitumor efficacy.
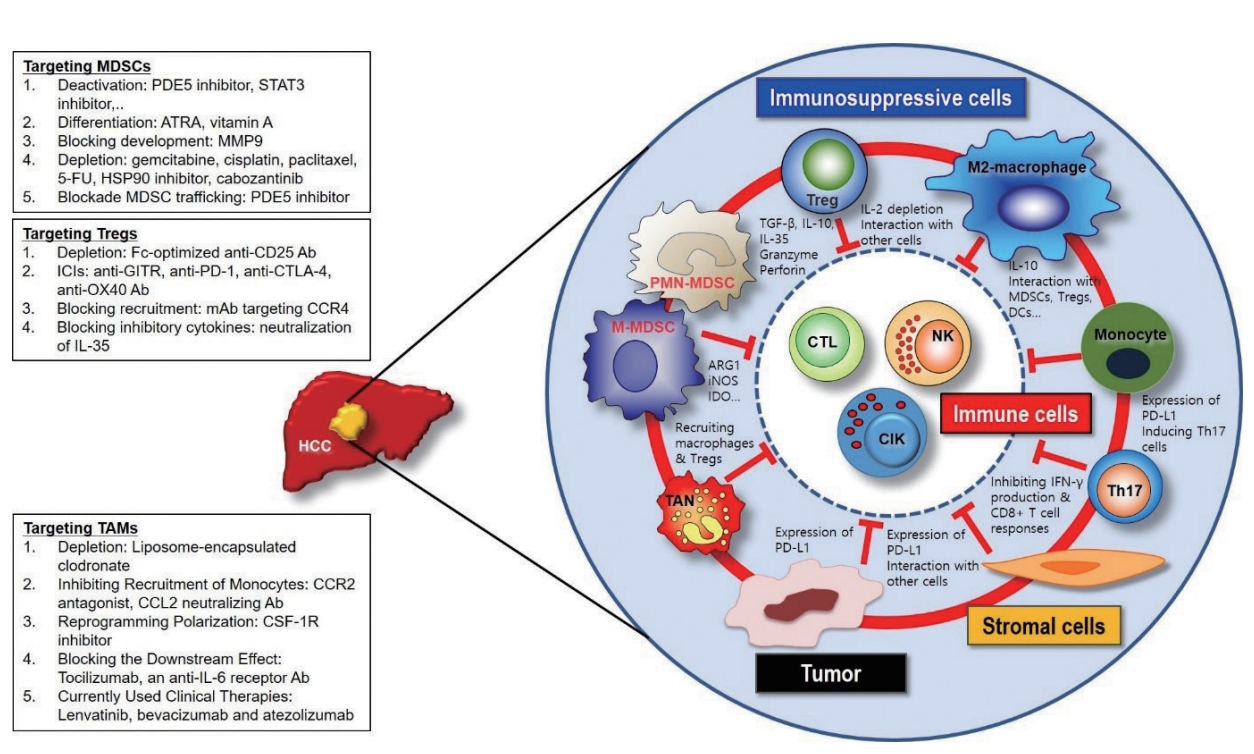
Cross-talks among immune cells, immunosuppressive cells, stromal cells, and tumor cells in the tumor microenvironment of hepatocellular carcinoma. PDE, phosphodiesterase; STAT, signal transducer and activator of transcription; ATRA, all-trans-retinoic acid; MMP, matrix metalloproteinase; FU, fluorouracil; HSP, heat shock protein; MDSCs, myeloid-derived suppressor cells; GITR, glucocorticoid-induced TNFR family related gene; PD, programmed cell death protein; CTLA, cytotoxic T-lymphocyte-associated protein; CCR, cell cycle-related kinase; IL, interleukin; TAM, tumor associated macrophages; CCL, C-C motif chemokine ligand; CSF, stimulating factor; Treg, regulatory T-cells; PMN, polymorphonuclear; TAN, tumor-associated neutrophils; TGF, transforming growth factor; ARG, arginase; iNOS, inducible nitric oxide synthase; IDO, indoleamine 2,3-dioxygenase; DC, dendritic cells; NK, natural killer; CIK, cytokine-induced killer; TNFR, tumor necrosis factor receptor.
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, NCI and in part by the Scientific Research Fund of the Korean Liver Cancer Study Group 2015.
Notes
The authors have no conflicts to disclose.